Line-ID
In this tutorial, we will identify species in a given spectrum and derive a quantitative description of their contributions using the LineID function, see Sect. “LineID”.
All files used within this tutorial can be downloaded here.
Observational xml file
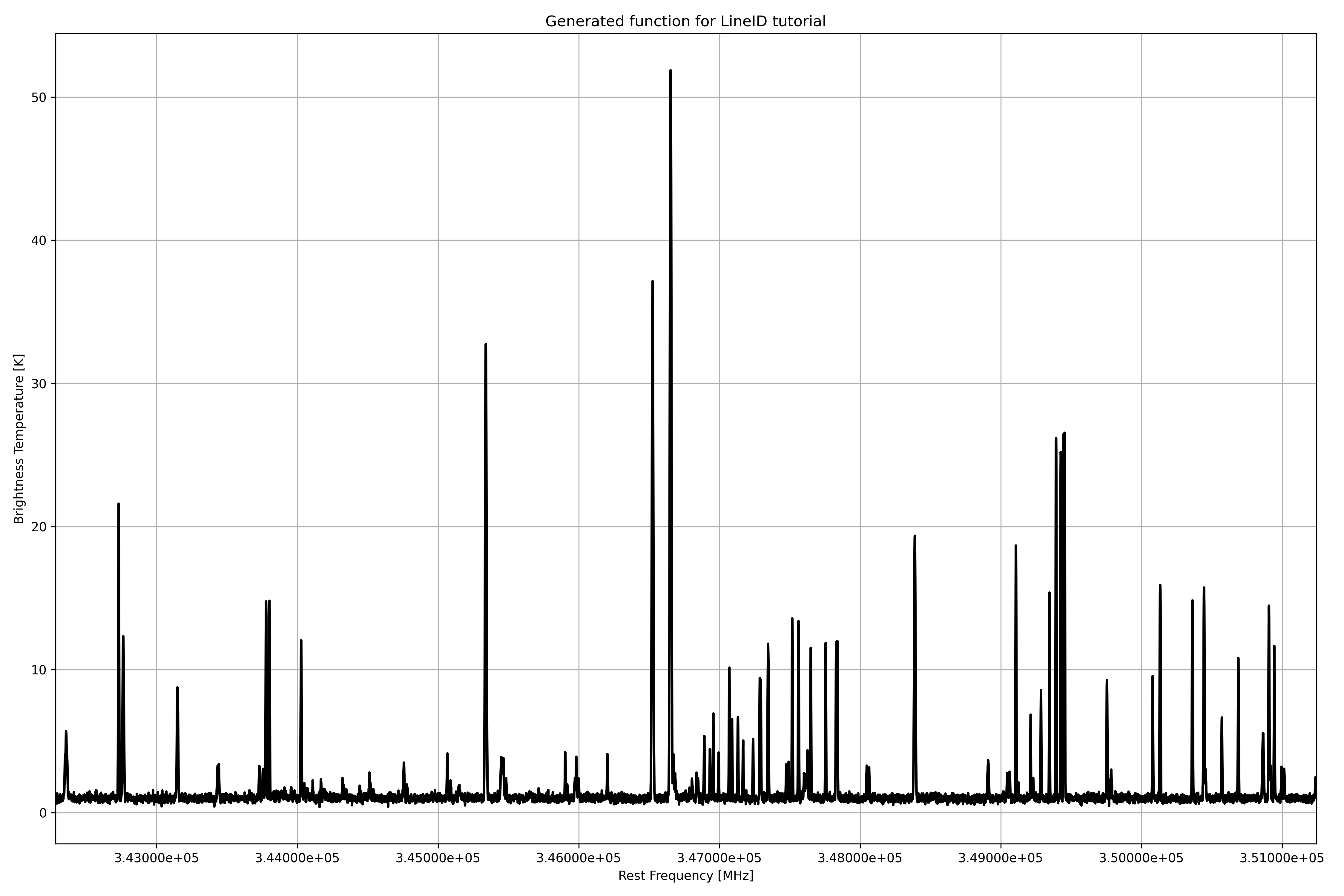
Fig. 24 The spectrum generated with the help of the myXCLASS function.
For the LineID function we have to define the frequency range, (here from 342.281 to 351.243 GHz), the size of the interferometric beam (here 0.4 arcsec), the background and dust parameters. Here, we use a synthetic spectrum, see Fig. 24, where we know, which molecules are included. Additionally, we add some white noise.
Observational xml file tutorial-lineid__obs.xml used for the
myXCLASSMapFit function:
<?xml version="1.0" encoding="UTF-8"?>
<ExpFiles>
<!-- define number of observation files -->
<NumberExpFiles>1</NumberExpFiles>
<!-- ************************************************************** -->
<!-- define parameters for observational data -->
<file>
<FileNamesExpFiles>files/data/SyntheticData.dat</FileNamesExpFiles>
<!-- define format -->
<ImportFilter>xclassASCII</ImportFilter>
<!-- define number of frequency ranges -->
<NumberExpRanges>1</NumberExpRanges>
<!-- define parameters for each data ranges -->
<FrequencyRange>
<MinExpRange>342281.0</MinExpRange>
<MaxExpRange>351243.0</MaxExpRange>
<StepFrequency>1.0</StepFrequency>
<!-- define background temperature and temperature slope -->
<t_back_flag>True</t_back_flag>
<BackgroundTemperature>1.0</BackgroundTemperature>
<TemperatureSlope>0.0</TemperatureSlope>
<!-- define hydrogen column density, beta for dust, and kappa -->
<HydrogenColumnDensity>1.4e+23</HydrogenColumnDensity>
<DustBeta>1.4</DustBeta>
<Kappa>0.0</Kappa>
<!-- define noise level for current frequency range -->
<NoiseLevel>0.5</NoiseLevel>
</FrequencyRange>
<!-- define local standard of rest (vLSR) -->
<GlobalvLSR>0.0</GlobalvLSR>
<!-- define size of telescope -->
<TelescopeSize>0.4</TelescopeSize>
<!-- define if interferrometric observation is modeled -->
<Inter_Flag>True</Inter_Flag>
<!-- define parameters for ASCII file import -->
<ErrorY>no</ErrorY>
<NumberHeaderLines>0</NumberHeaderLines>
<SeparatorColumns> </SeparatorColumns>
</file>
<!-- parameters for isotopologues -->
<iso_flag>False</iso_flag>
<IsoTableFileName> </IsoTableFileName>
</ExpFiles>
Molfit file
Now, we have to define a so-called default molfit file which defines for a certain molecule the number of components, the ranges and initial values for each parameter. During the line identification process the name of the (first) molecule defined in the default molfit file is replaced by the name of the current molecule.
The default molfit file tutorial-lineid__default.molfit used for the
LineID function:
%=======================================================================================================================================================================================================================================================================
%
% define database parameters:
%
%=======================================================================================================================================================================================================================================================================
%%MinNumTransitionsSQL = 1 % (min. number of transitions)
%%MaxNumTransitionsSQL = 90000 % (max. number of transitions)
%%MaxElowSQL = 8.000e+03 % (max. lower energy, i.e. upper limit of the lower energy of a transition)
%%MingASQL = 1.000e-09 % (minimal intensity, i.e. lower limit of gA of a transition)
%%TransOrderSQL = 1 % (order of transitions: (=1): by lower energy, (=2) by gA, (=3) gA/E_low^2, else trans. freq.)
%=======================================================================================================================================================================================================================================================================
%
% define molecules and their components:
%
%=======================================================================================================================================================================================================================================================================
SO2;v=0; 1
n 0.00 0.00 3.1E-01 y 3.00 900.00 80.00 y 1.e8 1.e23 1.00e+16 y 2.00 11.00 2.00 y -20.00 20.00 0.00 c
Iso ratio file
Here we do not use an iso-ratio file to keep the runtime short.
Algorithm xml file
In order to fit the parameters we used in the molfit file we have to define the
optimization algorithm(s), which is (are) used by the LineID function. Here,
we use the Levenberg-Marquardt algorithm, where we want to limit the number
of iterations per molecule to 10 by setting the tag
"<number_iterations>" to 10. Furthermore, we define another stopping criterion
by setting tag the upper limit of the \(\chi^2\) function to \(1 \cdot 10^{-6}\),
using the tag "<limit_of_chi2>". Additionally, we want to use two cores,
which is described by tag "<NumberProcessors>".
Algorithm xml file tutorial-lineid__algorithm.xml used for the
myXCLASSMapFit function:
<?xml version="1.0" encoding="UTF-8"?>
<FitControl>
<!-- settings for fit process -->
<!-- set number of used algorithms -->
<NumberOfFitAlgorithms>1</NumberOfFitAlgorithms>
<!-- define settings for 1st algorithm -->
<algorithm>
<FitAlgorithm>Levenberg-Marquardt</FitAlgorithm>
<!-- define value of the variation -->
<VariationValue>1.e-3</VariationValue>
<!-- set max. number of iterations -->
<number_iterations>10</number_iterations>
<!-- set max. number of processors -->
<NumberProcessors>8</NumberProcessors>
<!-- settings for chi^2 -->
<limit_of_chi2>1e-6</limit_of_chi2>
<RenormalizedChi2>yes</RenormalizedChi2>
<DeterminationChi2>default</DeterminationChi2>
<SaveChi2>yes</SaveChi2>
<!-- set plot options -->
<PlotAxisX>Rest Frequency [MHz]</PlotAxisX>
<PlotAxisY>T_mb [K]</PlotAxisY>
<PlotIteration>no</PlotIteration>
</algorithm>
</FitControl>
Call of LineID function
Now everything is prepared to start the LineID function.
Start LineID function
>>> from xclass import task_myXCLASSMapFit
>>> import os
# get path of current directory
>>> LocalPath = os.getcwd() + "/"
# import XCLASS packages
>>> from xclass import task_LineIdentification
# define path and name of default molfit file
>>> DefaultMolfitFile = LocalPath + "files/tutorial-lineid__default.molfit"
# define path and name of obs. xml file
>>> ObsXMLFileName = LocalPath + "files/tutorial-lineid__obs.xml"
# define list of molecules with are analyzed
>>> SelectedMolecules = ["CH3OCHO;v=0;", "CH3CHO;v=0;", "CH3OH;v=0;", \
"CH3CN;v=0;", "SO2;v=0;"]
# define upper limit of overestimation
>>> MaxOverestimationHeight = 500.0
# define tolerance
>>> Tolerance = 80.0
# define path and name of algorithm xml files
>>> AlgorithmXMLFileSMF = LocalPath + "files/tutorial-lineid__algorithm.xml"
>>> AlgorithmXMLFileOverAll = LocalPath + "files/tutorial-lineid__algorithm.xml"
# define lower limit for column density of core components
>>> MinColumnDensityEmis = 0.0
# define lower limit for column density of foreground components
>>> MinColumnDensityAbs = 0.0
# define source name
>>> SourceName = ""
# define list of so-called strong molecules
>>> StrongMoleculeList = []
## define path and name of cluster file
>>> clusterdef = ""
# call LineID function
>>> IdentifiedLines, JobDir = task_LineIdentification.LineIdentificationCore( \
MaxOverestimationHeight = MaxOverestimationHeight, \
SourceName = SourceName, \
DefaultMolfitFile = DefaultMolfitFile, \
Tolerance = Tolerance, \
SelectedMolecules = SelectedMolecules, \
StrongMoleculeList = StrongMoleculeList, \
MinColumnDensityEmis = MinColumnDensityEmis, \
MinColumnDensityAbs = MinColumnDensityAbs, \
AlgorithmXMLFileSMF = AlgorithmXMLFileSMF, \
AlgorithmXMLFileOverAll = AlgorithmXMLFileOverAll, \
experimentalData = ObsXMLFileName, \
clusterdef = clusterdef)
In general, the LineIdentification function considers all molecules which
have at least one transitions located within the frequency range(s) covered
by the obs. data file(s). Here, we use the parameter SelectedMolecules
to take only
CH\(_3\)OCHO\(_{v=0}\) ,
CH\(_3\)CHO\(_{v=0}\) ,
CH\(_3\)OH\(_{v=0}\) ,
CH\(_3\)CN\(_{v=0}\) , and
SO\(_{2, v=0}\) into account. Using the SQL parameters in the default
molfit file, it is possible to further restrict the transitions that are
analyzed by the LineID function.
Furthermore, we set the overestimation factor of the modeled single molecule
spectrum to 500 %. Please note, the input parameter MaxOverestimationHeight
has to be given in % and does not define the final overestimation limit
directly, which is given by MaxOverestimationHeight + 100 %.
If the modeled spectrum does not overestimate the observed spectrum, the corresponding molecule is “for now” identified and the fitted molfit file is added to the overall molfit file which is used at the end of the line identification process to determine the final contribution of each molecule.
The parameter Tolerance defines the max. fraction (in %) of overestimated
lines. If the fraction of overestimated lines in the result of a single
molecule fit is lower than the given threshold, the corresponding molecule
is “for now” identified and the optimized molfit file (and the corresponding
iso ratios) are considered in the final overall fit.
After finishing the single molecule fits, the LineID function performs a
final called overall fit taking all identified molecules into account, where
the fit parameters of all molecules are fitted simultaneously. The input parameter
AlgorithmXMLFileOverAll defines the algorithm(s), which is (are) used for
this fit.
Results
As described in Sect. “LineID”, the LineIdentification function
creates a subdirectory within the job directory called single-molecule_fits.
Within this directory the LineIdentification function creates subdirectories
for each molecule, which is taken into account. Here, the subdirectories
- CH3CHO_v=0_
- CH3CN_v=0_
- CH3OCHO_v=0_
- CH3OH_v=0_
- SO2_v=0_
contain all files for each single molecule fits, respectively. Additionally,
the subdirectories Intermediate_identified_molecules and
Non_identified_molecules contain molfit files describing the identified
and unidentified molecules, respectively. The LineID function generates two
log files, Identified_Molecules.dat and LineID.log, each describing
the identified molecules and the details of the identification procedure.
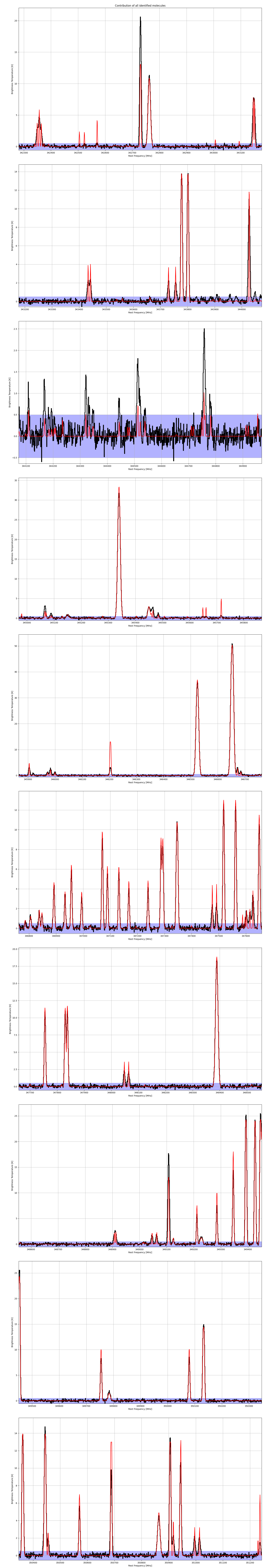
Fig. 25 The final spectrum with all identified molecules. The blue area around
the zero level represents the noise area, i.e. emission or absorption
features within this are ignored. The noise level is defined in the
obs. xml file, see above, by tag <NoiseLevel>.
Finally, the subdirectory all contains all files for the so-called
overall fit. In addition, the subdirectory final_fit is located
within the all directory as well, describing the contribution of all
identified molecules.